Understanding Friedreich’s Ataxia (FRDA)
Reduced expression of frataxin (Fxn) is the cause of Friedreich’s ataxia (FRDA), an early-onset neurodegenerative disease resulting in early mortality (median age of death, 35 years). Studies in mouse have shown that Fxn plays an important role during embryonic development (homozygous frataxin knockout mice display embryonic lethality). Existing transgenic and knockout FRDA animal models are designed to reduce frataxin either temporally, or selectively by targeting tissues that are severely affected in FRDA patients. These FRDA animal models are either mildly symptomatic due to insufficient reduction of frataxin (greater than 75%) or engineered to be tissue-specific conditional knockouts.
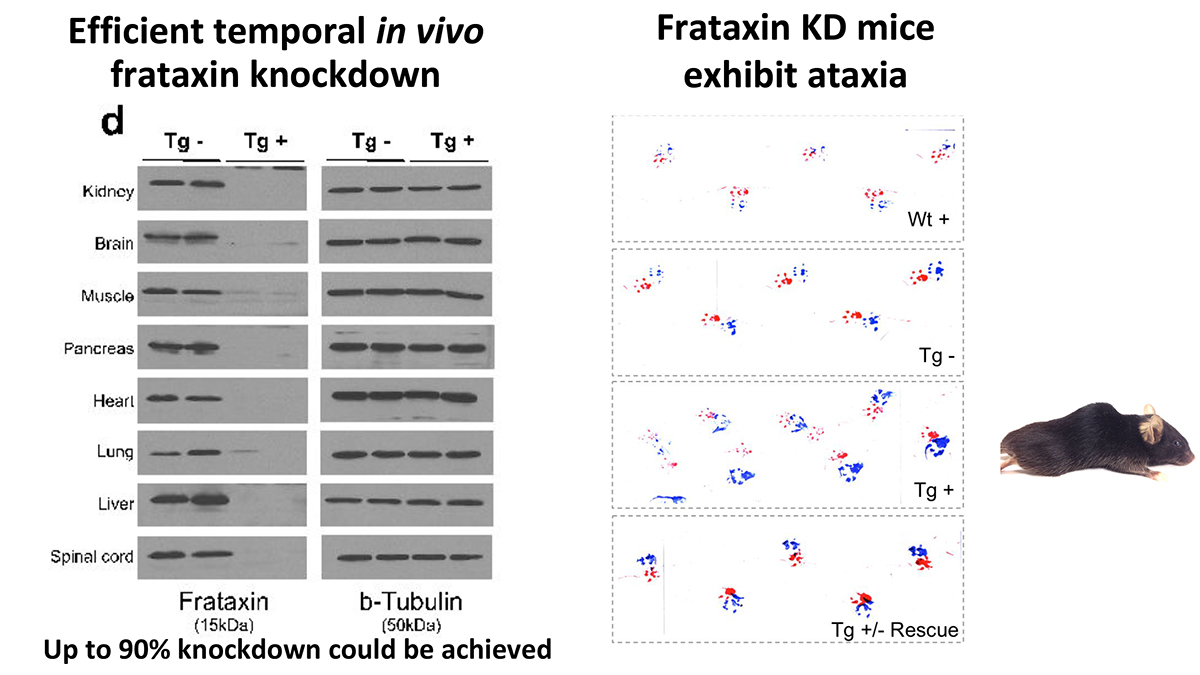
We have developed an inducible mouse model for FRDA that permits reversible frataxin knockdown and detailed studies of the temporal progression or recovery following restoration of frataxin expression. We targeted a single copy shRNA against the Fxn transgene (doxycycline-inducible) under the control of H1 promoter gene into the rosa26 genomic locus. This allowed us to circumvent the lethal effect of organism-wide knockout, while permitting significant frataxin reduction in all tissues. Fxn knockdown was achieved to control the onset and progression of the disease depending on the dose of doxycycline (Dox).

We found that systemic knockdown of Fxn in adult mice led to multiple features paralleling those observed in human patients, including electrophysiologic, cellular, biochemical and structural phenotypes associated with cardiomyopathy, as well as dorsal root ganglion and retinal neuronal degeneration and reduced axonal size and myelin sheath thickness in the spinal cord. Fxn knockdown mice also exhibited other abnormalities similar to patients, including weight loss, reduced locomotor activity, ataxia, reduced muscular strength, and reduced survival, as well as genome-wide transcriptome changes. The reversibility of knockdown also allowed us to determine to what extent observed phenotypes represent neurodegenerative cell death, or reversible cellular dysfunction. Remarkably, upon restoration of near wild-type Fxn levels, we observed significant recovery of function, pathology and associated transcriptomic changes, even after significant motor dysfunction was observed. This inducible model of FRDA is likely to be of broad utility in therapeutic development and in understanding the relative contribution of reversible cellular dysfunction to the devastating phenotypes observed in this condition.